Dada2 Pipeline Tutorial
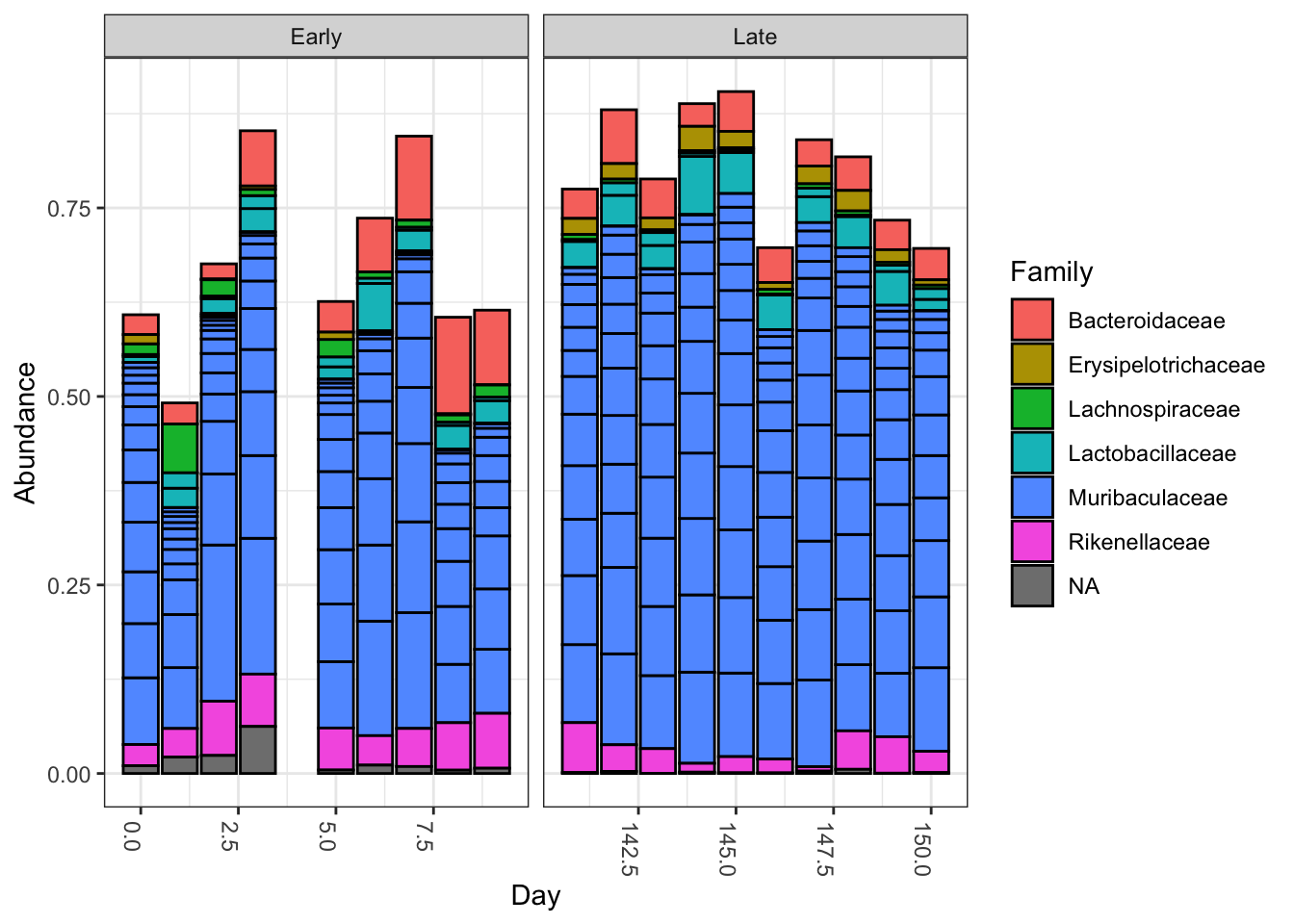
Well also include the small amount of metadata we have the samples are named by the gender G mouse subject number X and the day post-weaning Y it was sampled eg. Well also include the small amount of metadata we have the samples are named by the gender G mouse subject number X and the day post-weaning Y it was sampled eg.
Dada2 Pipeline Tutorial 1 16
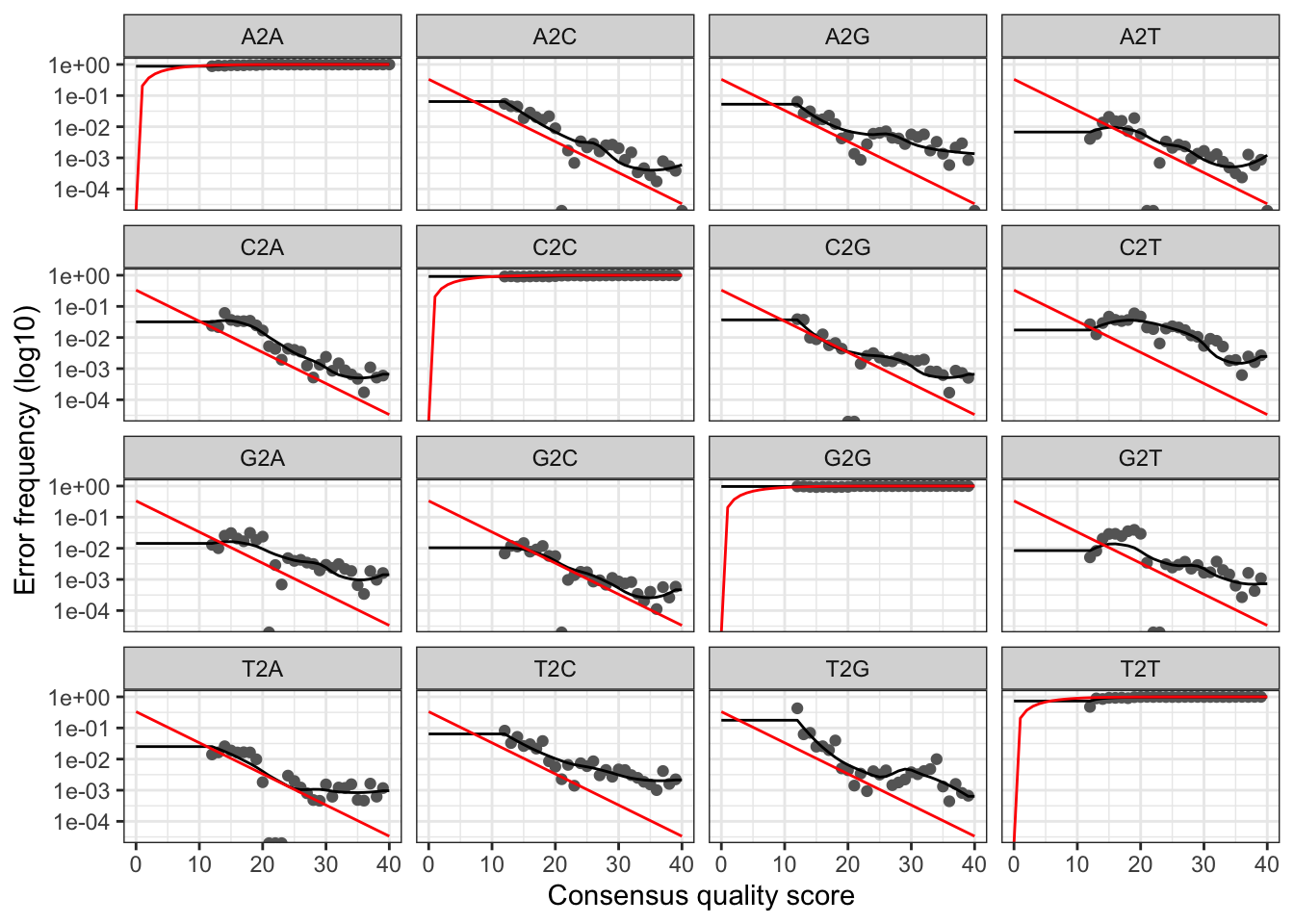
2 Overview of the dada2 pipeline.
Dada2 pipeline tutorial. 18th and 23rd of September 2019. The DADA2 pipeline produced a sequence table and a taxonomy table which is appropriate for further analysis in phyloseq. The dada2 R package manual.
The DADA2 pipeline is used as a method to correct errors that are introduced into sequencing data during amplicon sequencing. Then we perform some string manipulation to extract a list of the sample names. The DADA2 pipeline produced a sequence table and a taxonomy table which is appropriate for further analysis in phyloseq.
This tutorial created by Angela Oliverio and Hannah Holland-Moritz and updated May 13th 2019. The DADA2 output file is loaded file directly into the phyloseq workflow. 3 Overview of the dada2 pipeline.
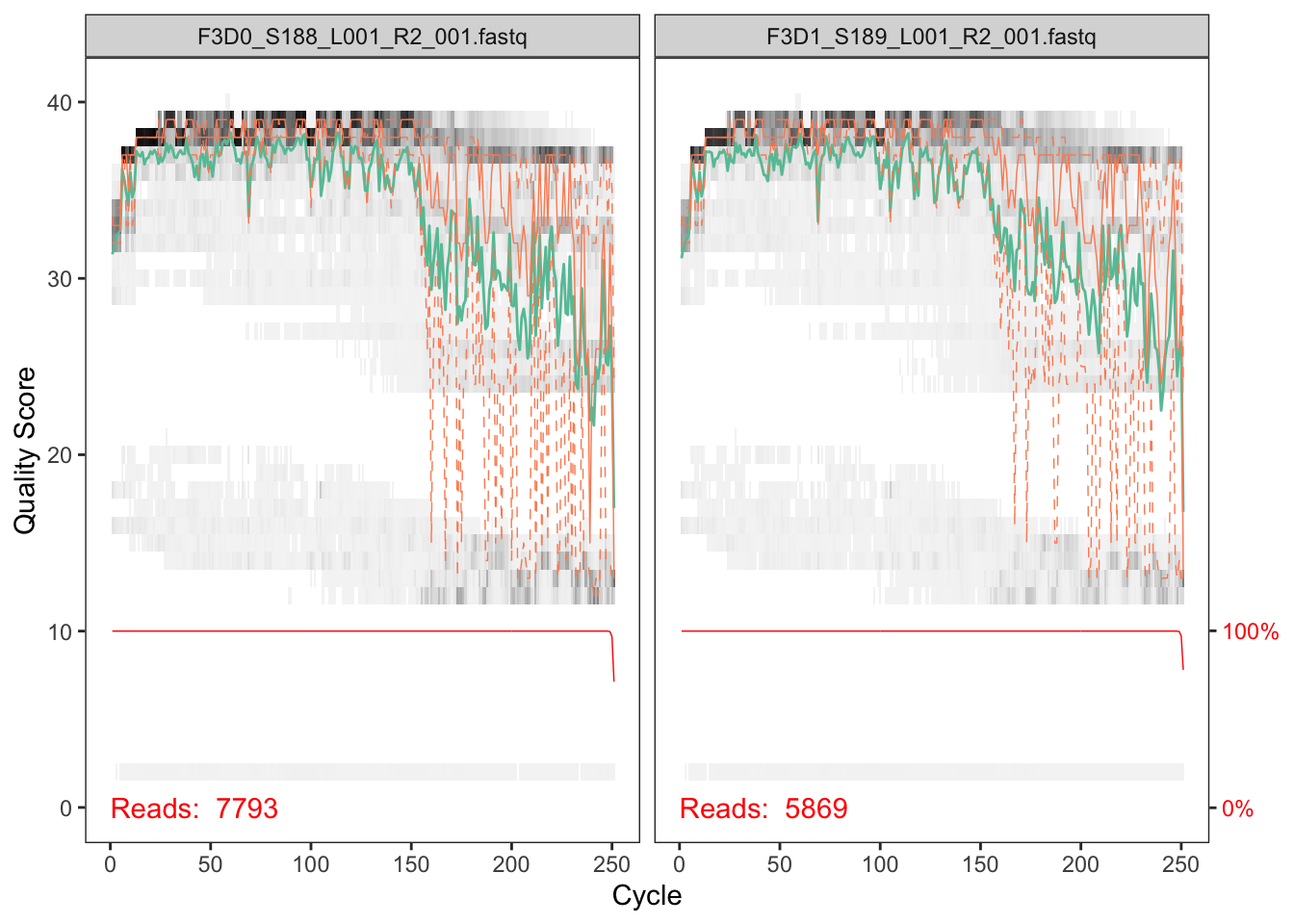
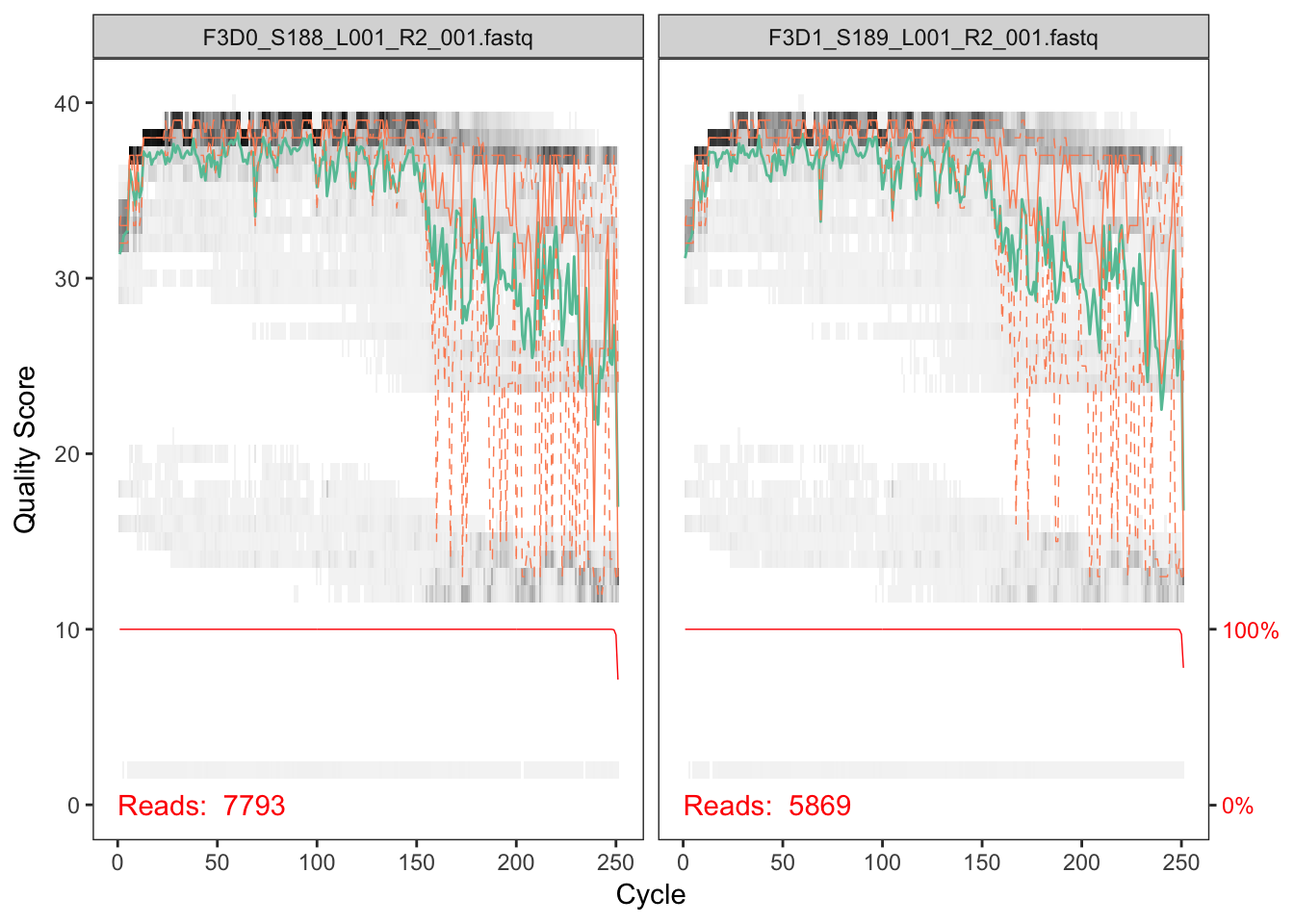
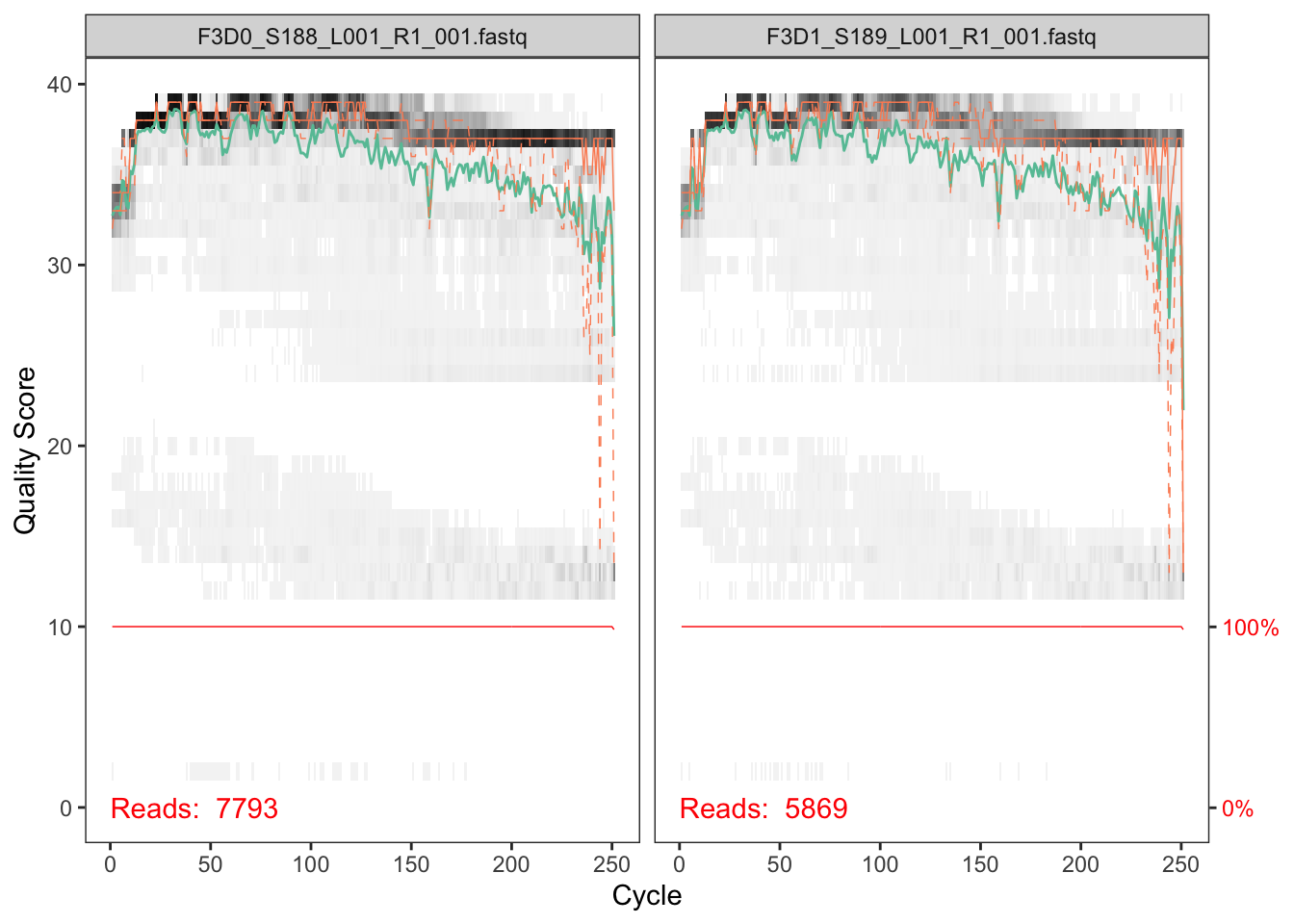
DOI for this workflow. Now we read in the names of the fastq files and we sort them by forward and reverse. This can serve as a jumping off point if youre left with too few sequences at the end to help point you towards where.
The developers DADA2 tutorial provides an example of a nice quick way to pull out how many reads were dropped at various points of the pipeline. The tutorial walkthrough of the DADA2 pipeline on paired end Illumina Miseq data. The starting point for the dada2 pipeline is a set of demultiplexed fastq files corresponding to the samples in your amplicon sequencing study.
Participants must have access to R or RStudio via their own computer or cluster. Discard reads that match against the phiX genome. Zachcp has definitely hit on some of the key points but this is the ideal large experiment pipeline as I currently see it.
TruncQ2Truncate reads at the first instance of a quality score less than or equal to truncQ keeping this as default. High resolution sample inference from Illumina. This pipeline runs the dada2 workflow for Big Data paired-end from Rstudio on the microbe server.
DADA2 Pipeline Tutorial 18 Here we walk through version 18 of the DADA2 pipeline on a small multi-sample dataset. We suggest opening the dada2 tutorial online to understand more about each step. The course is aimed at Master PhD students and other researchers working on microbiome studies.
If your listed files match those here you can start running the DADA2 pipeline. Sets the maximum number of expected errors allowed in a read which is a better filter than simply averaging. Using the following parameters.
There are many great tutorials and explanations out there on amplicon processing that you can dive into. This pipeline is exactly how we processed our data. That is dada2 expects there to be an individual fastq file for each sample or two fastq files one forward and one reverse for.
The dada2 pipeline takes as input demultiplexed fastq files and outputs the sequence variants and their sample-wise abundances after removing substitution and chimera errors. Running DADA2 on large experiments is something that can be done right now but needs its own tutorial. MaxN0 DADA2 requires no Ns.
It is implemented as an open-source R-package that will allow you to run through the entire pipeline including steps to filter. DADA2 Pipeline Tutorial 12 Here we walk through version 12 of the DADA2 pipeline on a small multi-sample dataset. The starting point for the dada2 pipeline is a set of demultiplexed fastq files corresponding to the samples in your amplicon sequencing study.
This is not meant to be a tutorial and we only provide minimal annotation. Dada2 tutorial with MiSeq dataset for Fierer Lab. That is dada2 expects there to be an individual fastq file for each sample or two fastq files one forward and one reverse for each sample.
Further documentation is available on the DADA2 front page. Our starting point is a set of Illumina-sequenced paired-end fastq files that have been split or demultiplexed by sample and from which the. Our starting point is a set of Illumina-sequenced paired-end fastq files that have been split or demultiplexed by sample and from which the.
This pipeline below is exactly how we processed our 16S rRNA data using DADA2 Callahan et al. This however is not meant to be a tutorial and we only provide minimal annotation. The material of taught in this module was developed by Benjamin Callahan.
Taxonomic classification is available via a native implementation of the.
Dada2 Pipeline Tutorial 1 16
Dada2 Pipeline Tutorial 1 16
Dada2 Pipeline Tutorial 1 8
Dada2 Pipeline Tutorial 1 16
{kind=link}